
EPITHELIAL BARRIER FUNCTION IN TRANSPLANTED HUMAN INTESTINAL ORGANOIDS IS IMPROVED BY ENTERIC NERVOUS SYSTEM BUT NOT EPITHELIAL DEVELOPMENTAL MATURITY
This session will provide the latest developmental research in human-derived organoids.
Methods: We generated colonoids from rectal biopsies obtained from pediatric patients with endoscopically active and inactive UC, and non-IBD controls [n=4/grp, median age 14.5 yrs]. Cryopreserved colonoids were thawed, passaged, and differentiated for 3 days. We analyzed the bioenergetics of colonoids (in quadruplicate) in real-time through Seahorse Analyzer MT stress testing. We used gene expression analysis, imaging, spectrometric, and flow cytometric methods to assess MT membrane potential (MMP), MT-reactive oxygen species (ROS) production, MT-mass, and MT-uncoupling proteins.
Results: At baseline, colonoids from active UC patients consumed more oxygen than those from non-IBD controls (P=.021), but produced a similar amount of ATP. Accordingly, we found an 85% increase in proton leak and lower MMP (assessed by JC-1 uptake) in active UC compared to non-IBD colonoids (P=.015) (Figure 1). Moreover, MT-ROS production was higher in active UC compared to non-IBD colonoids. In line with the observed higher proton leak in active UC, we found a significantly increased gene and protein expression of the MT respiratory chain uncoupling protein Ucp2 in active UC compared to non-IBD colonoids (P=.030 and .024, respectively, Figure 2). We validated the observed elevated Ucp2 in active UC through the analysis of rectal bulk RNA-seq data from the PROTECT Pediatric UC inception cohort (GSE109142). In the stem cell state before differentiation, MT-mass (normalized to 7-AAD- colonoid single cells) was similar among all colonoid groups. After differentiation, active UC colonoids exhibited higher mitochondrial mass than non-IBD controls (P=.019). Overall, colonoids from inactive UC patients showed similar metabolic characteristics to non-IBD controls.
Conclusion: Colonoids derived from active pediatric UC patients demonstrate inefficient MT bioenergetics marked by higher proton leak and the requirement of more oxygen to generate ATP compared to those from non-IBD patients. Such MT inefficiency may be caused by observed higher expression of uncoupling protein or increased ROS generation in UC colonoids. These data demonstrate that UC epithelial MT functional impairment can be modeled in patient-derived colonoids and support advancing colonoids as a preclinical human model for testing therapies against such metabolic dysfunction.

Figure 1: Impaired Mitochondrial metabolism in Active UC (UC) colonoids compared to non-IBD Control colonoids (C). (a) A sample well ready for Seahorse MitoStress Test after 3 d differentiation. Scale bar= 200 μm. (b) Bioenergetic profile of active UC vs C colonoids. (c) Increased basal respiration and (d) Elevated proton leak in active UC vs C colonoids.

Figure 2: Elevated uncoupling protein 2 (UCP2) expression in colonoids from active UC vs non-IBD controls. (a) Rectal bulk RNA-seq data from the PROTECT Pediatric UC inception cohort study showed a significant upregulation of Ucp2 gene in UC rectal tissues vs non-IBD controls. n=20 for C, 206 for UC. P < 0.0001 from the Mann-Whitney test. (b) Ucp2 mRNA is more abundant in UC colonoids compared to control. (c) Representative immunofluorescent images of OCT-embedded colonoids stained with DAPI (nuclei), Phalloidin (F-actin), mitochondrial marker antibody (Cox4), and Ucp2 antibody. Arrows show co-expression of Cox4+Ucp2+ (yellow marks) in the merged image. Scale bar = 50 μm. UCP2 expression was quantified in 4 high-power fields per sample and expressed per nucleated cells. n=4 patients/grp. The line within the boxplot represents the median, + represents the mean, and the symbol above and below the whiskers represents the outliers that are either greater than the 95th or less than the 5th percentile.
Methods: We determined hypoxia-induced molecular and functional responses in 3D human fetal intestinal organoids. Organoids were derived from human fetal intestinal tissue (18-21 weeks of gestation). Organoids were exposed to hypoxic (1% O2) and normoxic (21% O2) culture conditions for time periods up to 68 hours. Hypoxia-inducible factor signaling downstream target glucose transporter-1 (GLUT1) was used as marker for hypoxia-induced epithelial damage in organoids and validated in NEC tissue. Morphological changes of organoids induced by hypoxia were assessed by hematoxylin and eosin staining on paraffin sections. Epithelial permeability was determined by FITC-dextran 4 kDa (FD4) flux, monitored by mean fluorescence intensity ratios of lumen versus basolateral comportments. Differentially expressed genes were assessed by RNA-seq after 6 and 24 hours of hypoxia compared to normoxia. Apoptosis analysis were performed after hypoxia exposure. AnnexinV+ cell populations were determined by flow cytometry, and propidium iodide (PI) positive organoids were observed by fluorescence microscopy.
Results: Immunofluorescence microscopy showed 4.4 and 3.0 fold increase of relative GLUT1 expression after 6 and 24 hours of hypoxia exposure compared normoxia in organoids (n=2 donors). Increased levels of GLUT1 were observed along intestinal damaged areas in NEC tissue (n=2). Ruffled membranes, observed at 24 hours after hypoxia, indicated hypoxia-induced morphological changes in organoids. Compared to normoxia, hypoxia lead to increased FD4 flux at 6 and 24 hour time points (11% (p=0.09) and 59% (p=0.025) respectively, n=3). Amongst others, RNA-seq gene set enrichment analysis identified significantly enriched pathways (padj<0.05) related to hypoxia (incl. GLUT1), and apoptosis at 6 (n=5) and 24 hours (n=6) versus normoxia. AnnexinV analysis of post-hypoxia and control cell suspensions confirmed increased apoptosis after 24 and 68-72 hours (16% (p=0.07) and 27% (p=0.006) respectively; n=5). Also hypoxia-induced increased PI intensity levels were observed by microscopy.
Conclusions: The current results suggest that our human intestinal organoid-hypoxia model may be used as a proxy for hypoxia-induced intestinal damage in early life intestinal diseases. Our model could support identification of potential therapeutic targets that can be evaluated at epithelial functional and molecular level.
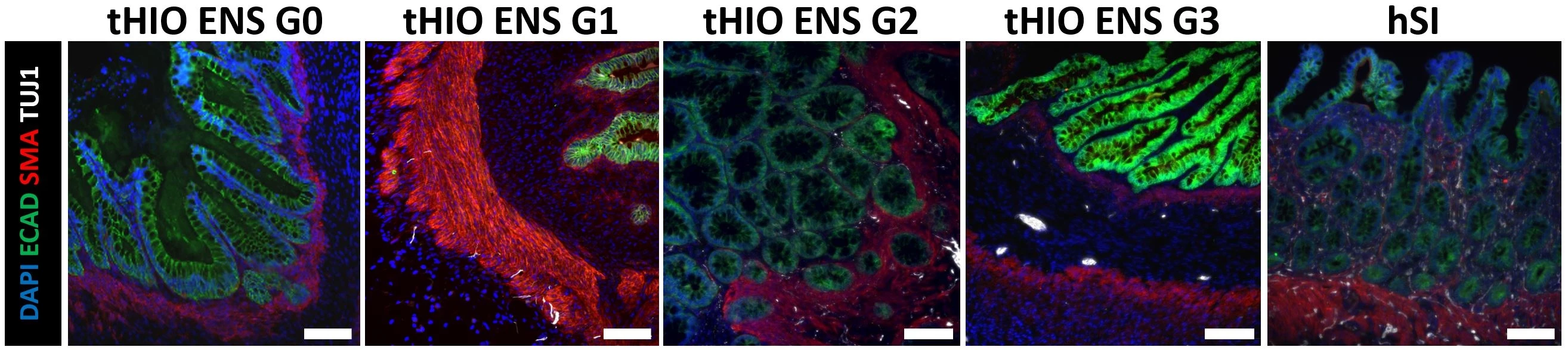
Methods: HIOs and ENCCs were generated from human embryonic stem cells (hESCs). HIOs and ENCCs were co-cultured in vitro for 28-40 days. HIOs±ENCCs were transplanted under the kidney capsule of adult NSG mice for up to 18 weeks. Transepithelial electrical resistance (TER) was recorded in an Ussing chamber as a measure of barrier function. H&E staining was performed to evaluate epithelial development per our lab’s previously described grading scale (G1 = absence of crypts/villi to G4 = elongated crypt-villus axis). Immunofluorescence (IF) staining was performed for epithelial marker (ECAD), smooth muscle actin marker (SMA), and neuronal marker (TUJ1), to evaluate ENS development. We developed an ENS grading scale from G0 to 3 with G0 = no TUJ1 present, G1 = paucity of TUJ1, G2 = ample TUJ1 but no ganglia, G3 = ample TUJ1 and ganglia present. tHIOs were compared to human small intestine (hSI) as controls (Figure 1).
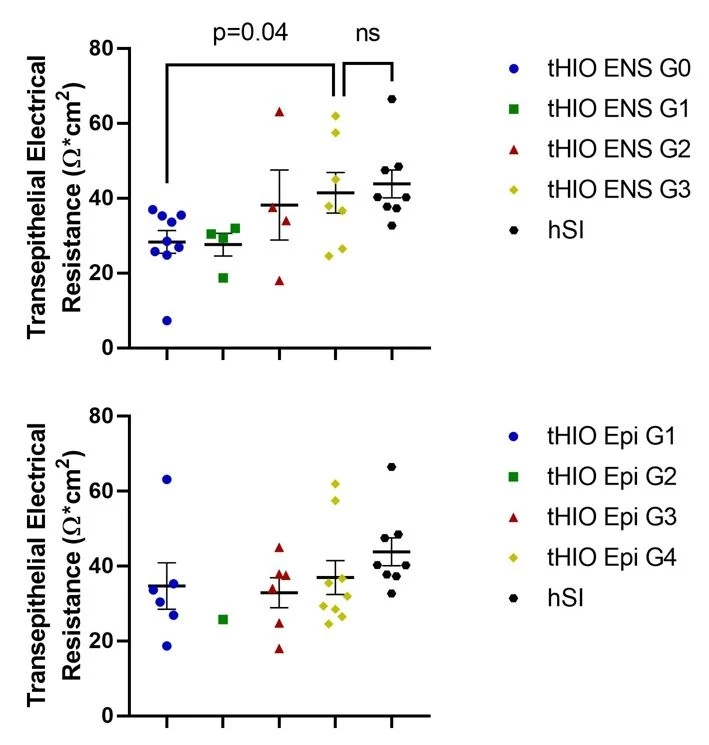
Results: H&E and IF staining confirmed that tHIOs matured in vivo with a robust epithelium (ECAD) surrounded by smooth muscle (SMA). IF also confirmed the presence of enteric neurons (TUJ1). TER was significantly higher in tHIOs with ENS G3 (41.5±5.4) versus ENS G0 (28.31±3.0) (p=0.04) and no different from hSI (43.8±3.7) (p=0.71). However, TER was not significantly different amongst the four epithelial developmental grades (Figure 2).
Conclusion: Epithelial barrier function in tHIOs is improved by ENS but not epithelial developmental maturity. Future studies will aim to enhance ENS developmental maturity within the tHIO model and additionally determine how ENS developmental maturity influences other functions in tHIOs, like motility and nutrient absorption.
Presenter

Speakers
Related Products

