

757 - A NOVEL ROLE OF HNF4A IN MICROSATELLITE STABILIZED COLORECTAL CANCER: PROMOTING METASTASIS OF MICROSATELLITE STABILIZED COLORECTAL CANCER BY UPREGULATING CXCL12 TO RECRUIT MDSC CELLS
The speakers in this session will provide insights into diverse molecular mechanisms of metaplasia, dysplasia and neoplasia across the esophagus, stomach, and colon.
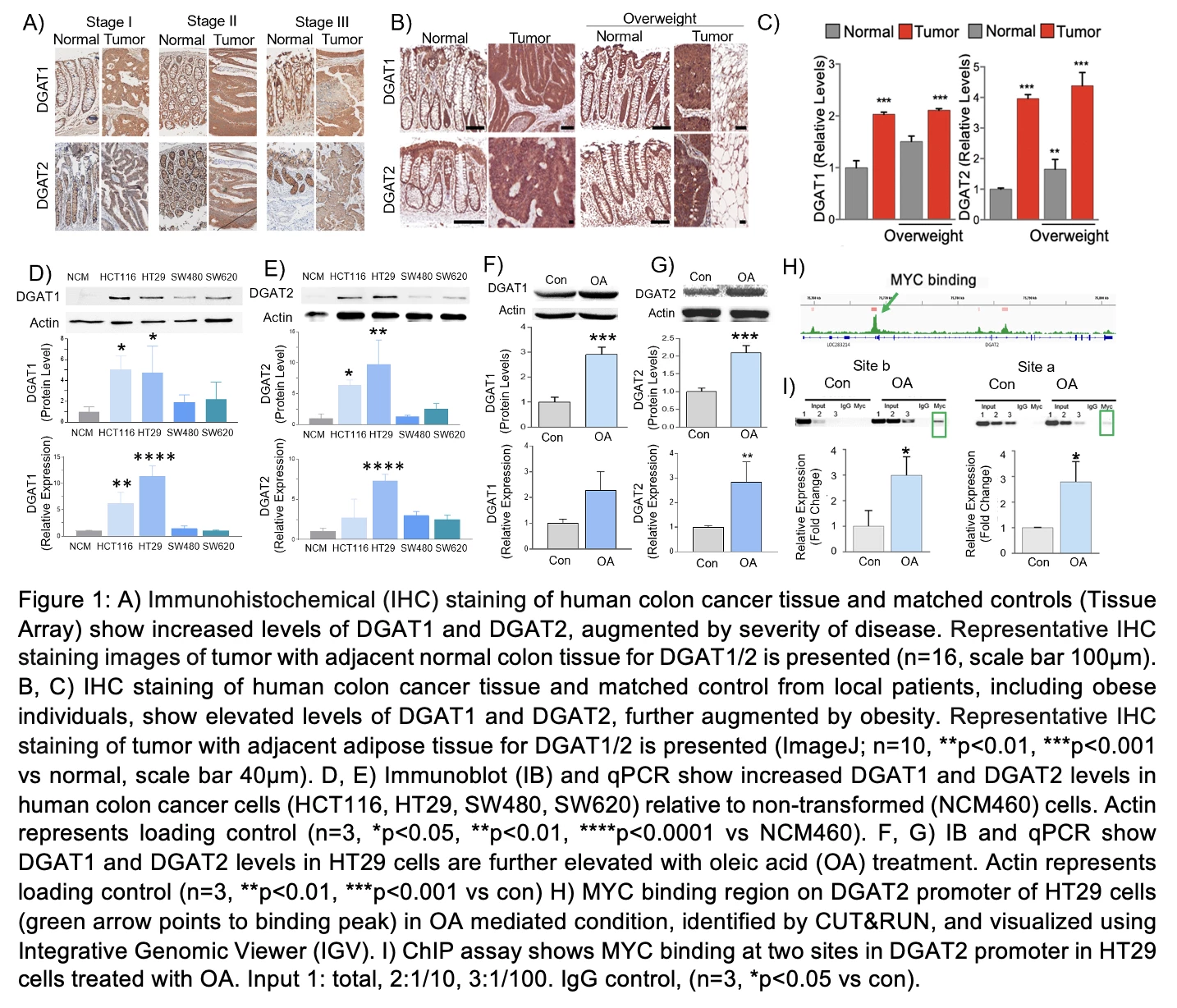
Figure 1A-I
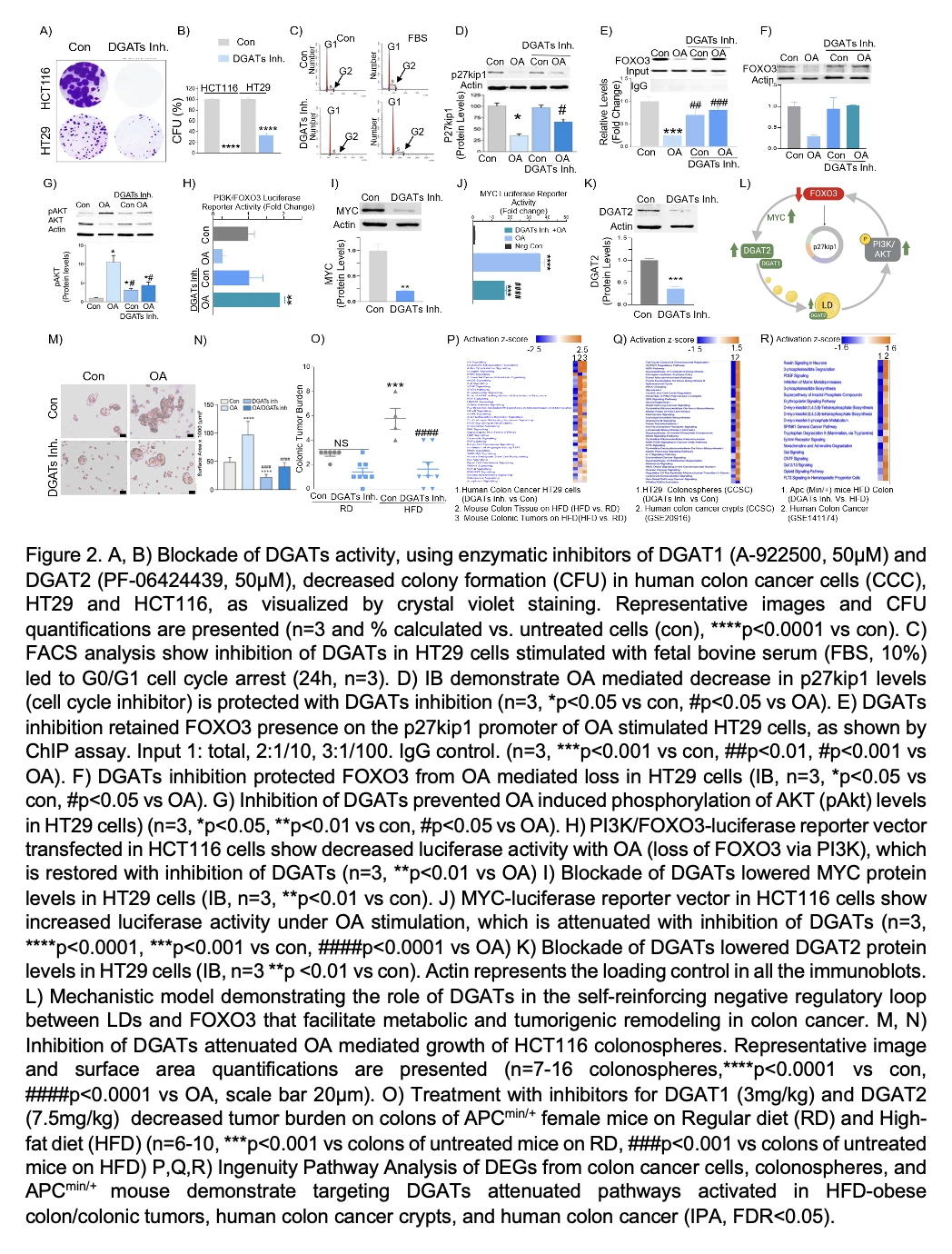
Figure 2A-R
Methods: HNF4A/CXCL12/TGFBI expression was examined by qRT-PCR, Western blot and IHC staining in MSS CRC tissues and cells. The clinical significance of HNF4A/CXCL12/TGFBI in two independent cohorts of CRC was assessed by KM analysis and the Multivariate Cox hazards model. The biological roles of HNF4A in tumor growth and metastasis were detected both in vivo and in vitro. The Inflammatory Cytokines & Receptors Profiler was used to explore the targets of HNF4A in MSS CRC. Serial deletion, site-directed mutagenesis luciferase report assays and chromatin immunoprecipitation were used to determine transcriptional regulation of CXCL12 promoters by HNF4A. MDSCs were isolated by flow cytometry and the Gr-1 monoclonal antibody was utilized to block the recruitment of MDSCs into tumors.
Results: HNF4A expression was upregulated in MSS CRC and negatively correlated with PDL1 expression. Upregulation of HNF4A promoted the proliferation and metastasis of MSS CRC cells and indicated poor prognosis. Meanwhile, HNF4A overexpression facilitates immunosuppression, with accumulated M0 and Tregs while reduced CD8+T, aNK, aDendritic, Monocytes and Neutrophils. The levels of HNF4A are positively correlated with the numbers of M0, Tregs and memory B cells, whereas negatively correlated with CD8+T, aNK, aDendritic, Neutrophils, Eosinophils and M1. Furthermore, CXCL12 was found to be the most upregulated cytokine upon HNF4A overexpression in MSS CRC cells. The proportion of MDSCs was remarkably enhanced in HNF4A-high CRC tissues of mice. While the use of Gr-1 antibody or CXCL12 knockdown rescued HNF4A-mediated CRC metastases. In addition, TGFBI/PI3K-AKT signaling was hyperactive in MSS CRC. TGFBI-induced HNF4A expression was blocked by PI3K inhibitor. The combination of the Gr-1 antibody and PI3K inhibitor attenuated HNF4A-mediated MSS CRC metastasis.
Conclusions: TGFBI-induced HNF4A overexpression promotes MSS CRC metastasis by transactivating the cytokine CXCL12 and thereby recruiting MDSC cells to suppress anti-tumor immunity in MSS CRC. The combination of Gr-1 antibody and PI3K inhibitor attenuated HNF4A-mediated MSS CRC metastasis, suggesting that HNF4A and CXCL12 as promising prognostic biomarkers and novel therapeutic targets for MSS CRC.

Figure 1. Upregulated HNF4A expression promoted the proliferation and metastasis of MSS CRC cells, predicting poor prognosis in CRC patients. (A-B)Differentially expressed genes between MSI and MSS CRC. (C-D)HNF4A expression was upregulated in MSS CRC and negatively correlated with PDL1 expression. (E-F)qRT-PCR and Immunohistochemistry staining analysis of HNF4A expression in adjacent non-tumorous and CRC tissues. (G-I)CRC patients with high HNF4A levels had an increased recurrence rate and decreased survival rate. HNF4A overexpression was positively correlated with lymphatic and distant metastasis of CRC. HNF4A upregulation was proved as an independent prognostic factor for CRC. (J-K)CCK-8 and transwell analysis of CRC cell proliferation and migration. (L-M) In vivo tumorigenesis and metastasis assay. Representative bioluminescent imaging, the overall survival, representative H&E staining of the liver and lung tissues, and the number of metastatic foci in different groups are shown.

Figure 2. TFGBI-induced HNF4A promotes metastasis of CRC by recruiting MDSC via CXCL12. (A-B)Upregulation of HNF4A facilitates immunosuppression, with increased M0 and Tregs while decreased CD8+T, aNK, aDendritic, Monocytes and Neutrophils in HNF4-high CRC. The levels of HNF4A are positively correlated with the numbers of M0, Tregs and mB cells, while negatively correlated with CD8+T, aNK, aDendritic, Neutrophils, Eosinophils and M1. (C)Cytokine microarray showed that CXCL12 was significantly upregulated upon HNF4A overexpression. The proportion of MDSC was remarkably enhanced in HNF4A-high CRC tissues of mice. (D-G)Representative BLI, tumor growth, overall survival, the number of metastatic foci and H&E staining in different groups are shown. (H)TGFBI was significantly upregulated in MSS CRC cells. (I)The PI3K-AKT pathway was hyperactive in MSS CRC. (J)TGFBI-induced HNF4A expression in a dose-dependent manner. (I)Blocking the PI3K-AKT pathway inhibits TGFBI-induced HNF4A expression.

